A rare hereditary disease called LAD-1 prevents patients’ immune cells from leaving the bloodstream, entering tissues, and fighting off infections. Therefore, unfortunately, most affected toddlers do not reach the age of two. An innovative gene therapy, grounded in a deep scientific understanding of the disease, now offers these children hope for leading healthy, full lives.
Advertisement
Rare hereditary diseases that affect as few as one newborn in a million pose a special challenge for patients and their families because so little information and research are available. These conditions, known as “orphan diseases,” receive scant attention from health systems and large pharmaceutical companies.
Nonetheless, small biotech firms—especially those working in gene therapy—often “adopt” orphan diseases and their patient communities. One reason is that a therapy for a rare disease with no existing treatment can obtain accelerated approval from regulatory authorities, giving a young company an their first "win". For this reason, we have recently seen a growing number of companies developing gene therapies for rare disorders. These treatments aim to correct the genetic defects that cause hereditary diseases, offering new—and sometimes the only—hope to people with orphan diseases and their families.
One such disease is leukocyte adhesion deficiency type 1, or LAD-1, characterized by the immune system’s inability to fight infections. The disorder is caused by mutations in the ITGB2 gene, which contains the instructions for producing an integrin protein whose crucial role we will describe below [1-3].
To protect the body from infections caused by bacteria or viruses, white blood cells must arrive at the affected tissue in time and eliminate the invaders. The bloodstream lets them travel, but aimless drifting will not achieve the goal; the cells must exit the blood vessel at the precise location that allows them to reach the afflicted tissue that is “calling for help.”
This impressive process has multiple steps: First, changes in the blood vessels around the afflicted site cause the white blood cell to slow down and make it stick to the vessel wall. When it detects molecules released from the infection site, the integrin proteins on its surface—acting like arms—activate and bind to receptors on the cells lining the vessel. Once anchored at the right spot, the white blood cell can squeeze out of the vessel (in a process termed diapedesis) and migrate to the infected tissue.
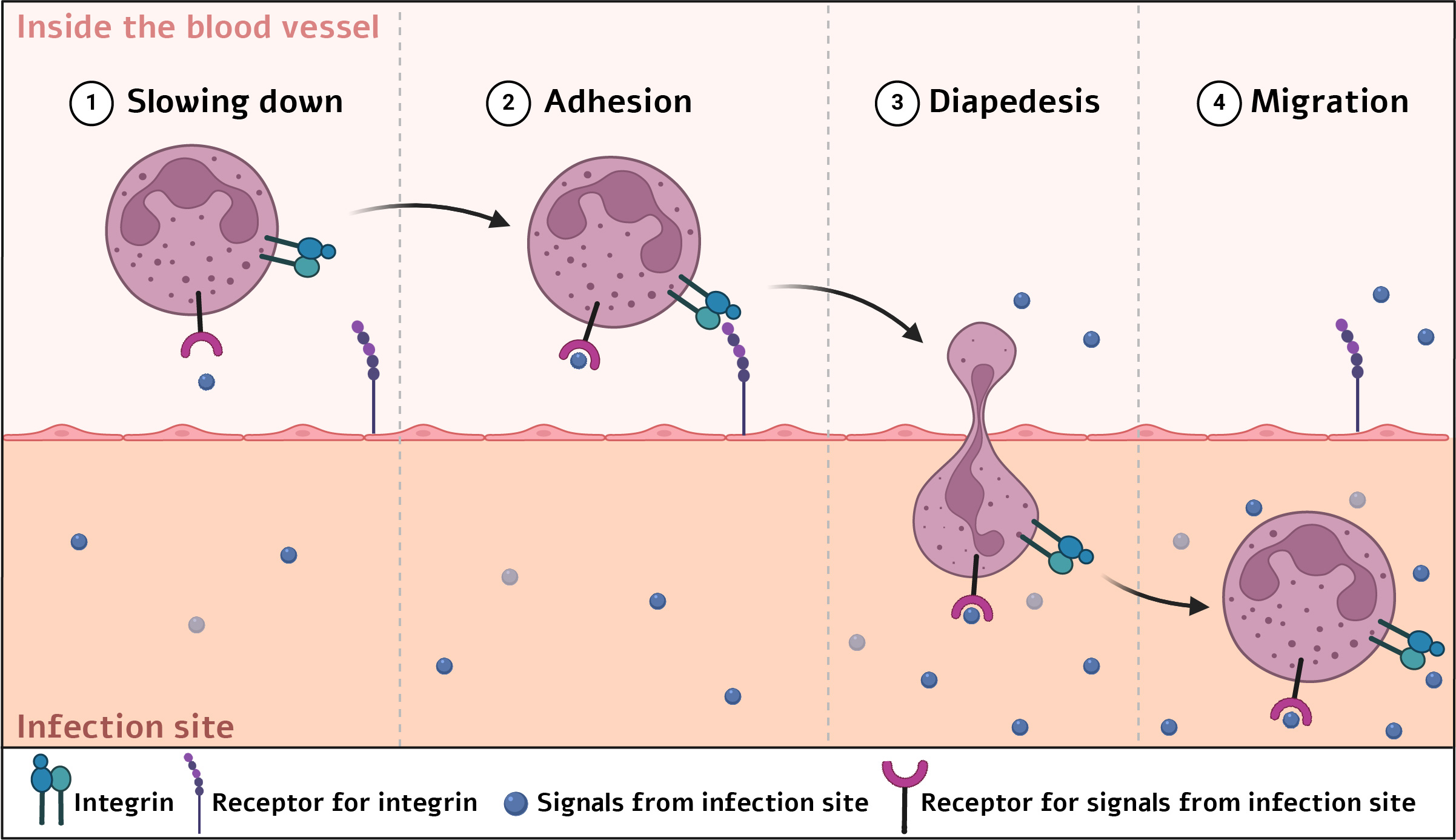
Steps by which immune cells exit blood vessels toward an infection site | Illustration: Elee Shimshoni via BioRender.com
A simple blood test on an LAD-1 patient gives us a hint into the problem: these infants have an excess of white blood cells in circulation. Without integrin, the cells are trapped in the blood vessels and cannot reach the places where they are needed. One hallmark of the disease is wounds and cuts that do not heal properly and lack pus, indicating that the normal and desired process by which neutrophils arrive to attack disease-causing agents is not working.
Due to the integrin deficiency, affected infants develop many severe infections in various body sites—such as the brain, lower airways, and gums—and spend most of their early childhood in repeated hospitalizations. The outlook is grim: more than 60% of affected children die before their second birthday.
Until now, the only life-saving option was a bone-marrow transplant from a donor. Bone marrow contains blood stem cells that can give rise to all blood cell types, including immune cells. A successful transplant means that the donor’s stem cells engraft in the patient’s marrow, multiply, and within a few months spread through the body, replacing the recipient’s immune system.
The initial hurdle is finding a compatible donor to reduce the risk that the donor’s immune cells will attack the patient’s body, a life-threatening condition called graft-versus-host disease. If a suitable donor is found, the process can begin. In LAD-1, only 58% of children who received a bone-marrow transplant from a donor survived the first three years without complications.
A new clinical study tested an innovative approach in nine children: inserting a healthy copy of the defective gene into the patients’ own blood-forming stem cells [4-5]. This strategy not only eliminates the need to find a genetically matched donor—saving precious time—but also removes the risk of graft-versus-host disease.
First, stem cells were harvested from the patients’ bone marrow. Next, the researchers exposed the cells to a genetically engineered virus that delivered a functional copy of the integrin gene into the cells’ genome. The virus was modified so it could not replicate further or infect additional cells.
As with a donor transplant, the patients then received chemotherapy to eradicate their own blood-forming stem cells, allowing the corrected cells to replace their immune system. Only after this step were the engineered cells infused back into the patients.
The results were striking: all nine children in the study survived, and at the time of publication, for at least two years [4]. Severe infections requiring hospitalization or intravenous treatment fell by about 85%. Moreover, wounds, ulcers, and cuts that had plagued the infants began to heal normally after therapy.
All reported side effects were linked to the chemotherapy given before infusion of the engineered cells, not to the therapy itself. Importantly, although the study showed a favorable safety profile, in gene therapies of this kind the healthy gene integrates randomly into the patient’s stem-cell genome, raising the possibility of insertion into problematic sites that could trigger cancer. The researchers found no evidence of this in any participant, but such an event could still occur in future patients.
The treatment, submitted for approval to the U.S. Food and Drug Administration (FDA), places LAD-1 among the small group of lethal hereditary blood disorders that have become treatable thanks to remarkable advances in biology and medicine [6-7]. Saving these infants requires deep understanding of the disease at both the genetic and clinical levels, combined with advanced molecular technologies that allow insertion of a healthy gene copy and safe reinfusion of the corrected cells. Such therapies, which can receive relatively rapid approval, offer hope to many families longing for a similar solution for their children with orphan diseases.
In the main image: three siblings with LAD-1 successfully treated with gene therapy | Courtesy of the family and the University of California, Los Angeles
Hebrew editing: Galia Halevy-Sarah
English editing: Elee Shimhsoni
References:
- Explanation of the disease from the Immune Deficiency Foundation
- Review article on the disease
- Additional review article on the disease
- Original paper describing the gene-therapy clinical trial for LAD-1
- Website of the company developing the gene therapy
- Story of three siblings treated with gene therapy
- Gene therapy for “bubble boy” disease
- First CRISPR-based therapy approved for sickle-cell anemia and β-thalassemia

Sagie holds a PhD from the Department of Molecular Genetics at the Weizmann Institute and works as a computational biologist at a company developing personalized cancer treatments. Scientific editor at "Little, Big Science".
Elee serves as Scientific Director at “Little, Big Science.” She holds a PhD in Biology from the Weizmann Institute and completed her postdoctoral training at Harvard and MIT. Her expertise lies in cancer, inflammation and metabolism, as well as in developing complex human-cell based model systems of disease.